Automated chemical reaction prediction: now in stereo
Research Press Release | December 01, 2022
Automated reaction path search method predicts accurate stereochemistry of pericyclic reactions using only target molecule structure.
The AFIR method traces back the reaction of endiandric acid C methyl ester, a 52-atom natural product, to its starting materials using only quantum chemical calculations. (Tsuyoshi Mita et al. JACS. November 30, 2022)
Researchers at the Institute for Chemical Reaction Design and Discovery (WPI-ICReDD) have demonstrated the expanded use of a computational method called the Artificial Force Induced Reaction (AFIR) method, predicting pericyclic reactions with accurate stereoselectivity based only on information about the target product molecule. The accurate prediction of a molecule’s stereochemistry—i.e., the 3D arrangement of its constituent atoms—is unprecedented for such an automated reaction path search method. This study serves as proof of concept that the AFIR method has the potential to discover novel reactions with specific stereochemistry.
In this study, AFIR is used to calculate retrosynthetic, or reverse, reactions going from product molecules to starting materials. Previously, AFIR has been used to predict small, simple reactions, but accurate stereochemistry predictions were out of reach, limiting the technique’s applicability. In this study, researchers overcome this hurdle by using the AFIR method on a major class of chemical reactions called pericyclic reactions, which are commonly found in biological processes, including the synthesis of Vitamin D.
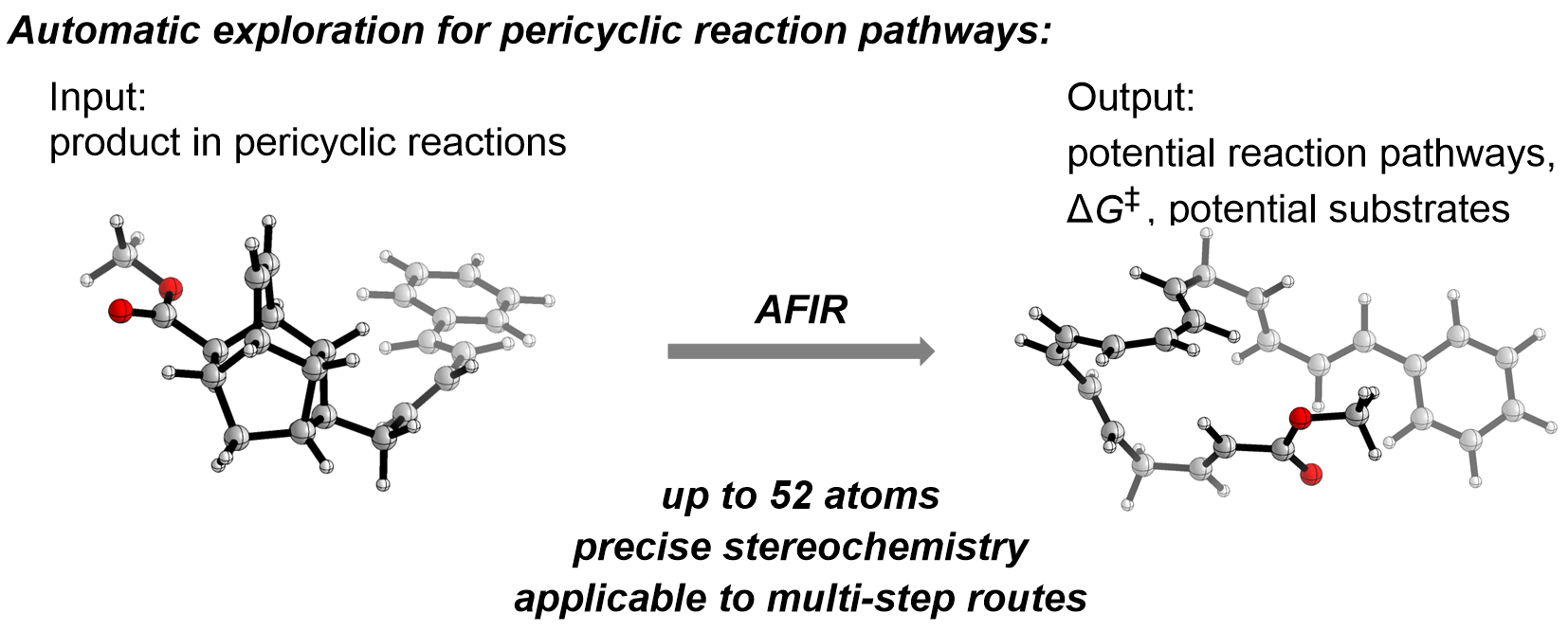
General scheme for generating retrosynthetic reaction path networks using the AFIR method. (Tsuyoshi Mita et al. Journal of the American Chemical Society. November 30, 2022)
A key property of pericyclic reactions is that they have the same stereochemical relationship whether the reaction is happening in the forward or backward direction. This enabled the team to use the stereochemistry computed for the backward reaction to accurately predict the stereochemistry for the forward reaction. Notably, AFIR was also able to correctly predict stereochemistry for a reaction that breaks the Woodward-Hoffman rules, which typically govern the behavior of pericyclic reactions. The capability of this technique to predict exceptions to these standard rules demonstrates the potential for using this automated method to discover unintuitive reactions that may otherwise be overlooked.
Pericyclic reactions are also advantageous for computational work because they are concerted, which means all the bond-breaking and bond-making happens in a single step. Since such reactions do not go through ionic intermediates, it is tough for solvent molecules to alter the reaction and allows researchers to ignore solvent effects in their computations. Such computations are simpler to perform and thus enable more complex molecules to be handled. In this study, AFIR successfully handled molecules of up to 52 atoms, over 2.5 times the size of molecules in previous studies.
Importantly, the only information needed to use AFIR is the structure of the desired product molecule, so researchers can input a molecule they want to make and effectively hit the rewind button to find promising starting materials. Expanding the use of AFIR to encompass larger molecules and stereospecific processes opens up new avenues for automated reaction discovery.
Lead author Tsuyoshi Mita was also struck by the significance of using an automated approach to recreate historically significant reactions.
“The Woodward-Hoffmann Rules were established in the 1960s and I think it is very profound that 60 years later we are able to use automated reaction path searching methods to predict a reaction’s starting materials with stereochemistry that follows these rules,” said lead author Tsuyoshi Mita. “Endiandric Acid C was synthesized by Nicolau in 1982 based on Black’s biosynthetic hypothesis, and I am very pleased that in this work we were able to recreate what was in their heads using quantum chemical computations. I am both an organic synthetic chemist and a user of the AFIR method and in this project I was once again reminded of the power of the AFIR method.”

Members of the research team at WPI-ICReDD, Hokkaido University. Top Row, Left to Right: Yu Harabuchi, Wataru Kanna, Hideaki Takano, Hiroki Hayashi. Bottom row, left to right: Satoshi Maeda, Tsuyoshi Mita. (Photo Credit: ICReDD)
Original Article:
Tsuyoshi Mita, Hideaki Takano, Hiroki Hayashi, Wataru Kanna, Yu Harabuchi, Kendall N. Houk, and Satoshi Maeda. Prediction of High-Yielding Single-Step or Cascade Pericyclic Reactions for the Synthesis of Complex Synthetic Targets. Journal of the American Chemical Society. November 30, 2022.
DOI: 10.1021/jacs.2c09830
Funding:
This work was financially supported by the Japan Science and Technology Agency Exploratory Research for Advanced Technology (JST-ERATO) grant (JPMJER1903), Japan Society for the Promotion of Science (JSPS) World Premier Initiative (WPI), and JSPS Grants-in-Aid for Challenging Research (Exploratory) (21K18945), Scientific Research (B) (22H02069), Transformative Research Areas (A) (Digitalization-driven Transformative Organic Synthesis (Digi-TOS)) (22H05330), and Young Scientists (22K14673). It was also supported by the Fugaku Trust for Medical Research, the Uehara Memorial Foundation, the Naito Foundation and the US National Science Foundation (CHE-1764328). All calculations were performed using the supercomputer Fugaku provided by the RIKEN Center for Computational Science as well as the supercomputer system at the Information Initiative Center, Hokkaido University, Sapporo, Japan. The use of the GRRM and Gaussian programs on the supercomputer Fugaku was enabled with the support of HPC Systems, Inc.
Contacts:
Specially Appointed Associate Professor Tsuyoshi Mita
Institute for Chemical Reaction Design and Discovery (WPI-ICReDD); Faculty of Engineering
Hokkaido University
Email: tmita[at]icredd.hokudai.ac.jp
Professor Satoshi Maeda
Institute for Chemical Reaction Design and Discovery (WPI-ICReDD); Faculty of Engineering
Hokkaido University
Email: smaeda[at]eis.hokudai.ac.jp
Collin Stecker (Public Relations and Outreach)
Institute for Chemical Reaction Design and Discovery (WPI-ICReDD)
Hokkaido University
Tel: +81-11-706-9646
E-mail: public_relations[at]icredd.hokudai.ac.jp
Sohail Keegan Pinto (International Public Relations Specialist)
Public Relations Division
Hokkaido University
Tel: +81-11-706-2185
Email: en-press[at]general.hokudai.ac.jp
Related Press Releases:
Chemical reaction design goes virtual
Novel computer-assisted chemical synthesis method cuts research time and cost | Hokkaido University