How the African clawed frog got an extra pair of genes: Whole genome sequence reveals evolutionary history of Xenopus laevis
Research Press Release | October 24, 2016
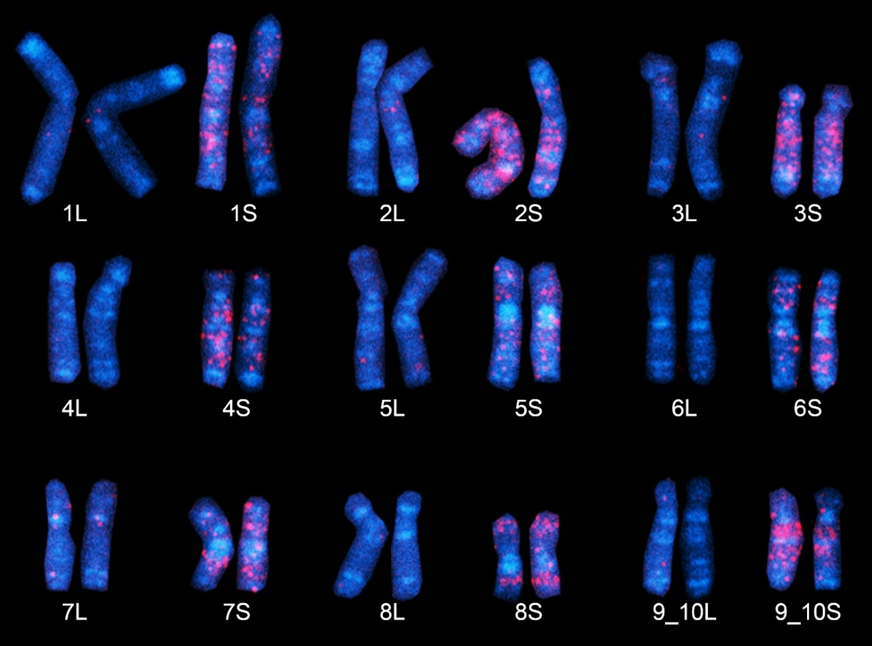
| African clawed frog chromosomes. “Fossil” DNA sequences found only in the S subgenome (chromosomes) of Xenopus laevis emit a red glow employing the FISH method. The nine S chromosomes here clearly possess a larger number of red regions, believed to have derived from ancestral species S. (Chromosome 9, at bottom right, is represented as “9_10,” as it corresponds to the fusion of ancestral chromosomes corresponding to chromosomes 9 and 10 in Xenopus tropicalis). Copyright: 2016 Yoshinobu Uno. |
The African clawed frog’s ancestor inherited one set of chromosomes each from two different species and doubled its whole genome some 18 million years ago, according to an international research consortium led by Japanese and American scientists who sequenced the entire genome of the Xenopus laevis for the first time. Scientists hope that the finding will help our understanding of vertebrate evolution, as the vertebrate genome doubled twice 500 million years ago.
Xenopus laevis is unusual in that it is a tetraploid species that has four sets of chromosomes, while many organisms, including humans, are diploid and have two sets of chromosomes. How and when this came about has been a topic of debate for some time.

| African clawed frog (right) and Western clawed frog. Though similar in appearance, the tetraploid Xenopus laevis adult female is noticeably larger than its counterpart in the related diploid species Xenopus tropicalis. Copyright: 2016 Shuji Takahashi. |
One hypothesis is that the tetraploid X. laevis inherited one half of its genetic material from each parent when two diploid ancestral species mated, and the genome of this diploid offspring then doubled, giving rise to a tetraploid organism with twice the number of chromosomes as its ancestors.
Xenopus laevis is an essential organism for biological and biomedical research, but the sheer size and complexity of its genome made it difficult for scientists to sequence the genome in its entirety. Sequencing the entire genome would not only be valuable for biological and biomedical research but also provide clues as to the origins of tetraploidy.
The researchers sequenced the entire genome of the J (Japan) strain of the frog, which was developed by Hokkaido University scientists, who inbred the animal over 20 years. The strain is genetically homogeneous, giving scientists a big advantage in the sequencing.
The US team, led by Dan Rokhsar and Richard Harland at the University of California, Berkeley, used the shotgun method to sequence short fragments of DNA and piece them together like a puzzle. The Japanese team, led by Masanori Taira at the University of Tokyo, sequenced very long fragments of DNA and determined the location of the long DNA on the chromosomes. Akimasa Fukui and his colleagues at Hokkaido University contributed particularly to data analysis and chromosome mapping. This extra step helped to distinguish the separate genome sequences inherited from each ancestral species.
It was a challenging idea to analyze the “transposable elements” (segments of DNA that move around the genome) that become fixed or inactive over time, but might be one way of tracing the two ancestral genomes, present as subgenomes in X. laevis. Akira Hikosaka at Hiroshima University and Yoshinobu Uno at Nagoya University (the latter an alumnus of Hokkaido University), tested this idea and discovered that indeed two sets of chromosomes originated from different diploid ancestors.
Curiously, the scientists also found that the subgenomes evolved separately in the nucleus, giving rise to shorter S- and longer L-types, the first evidence of animal subgenome evolution. The L-type chromosomes preserved most of the ancestral genetic information, while the S-type showed greater gene loss, deletion and rearrangement. Furthermore, it was suggested that any number of genes were just changing their functions.
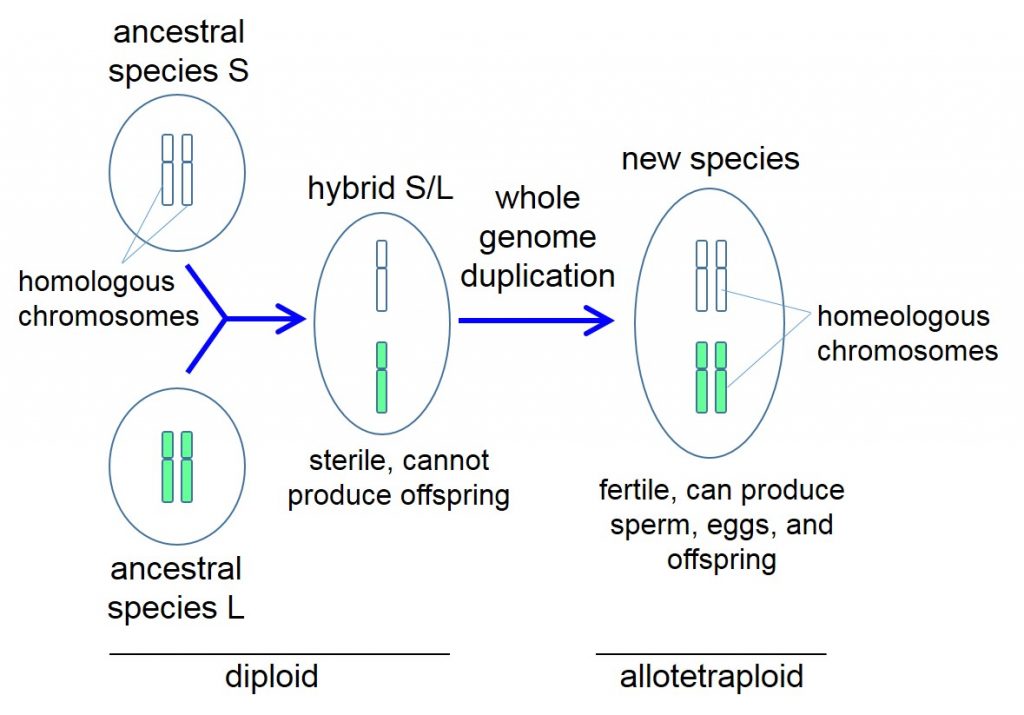
| Genesis of the Xenopus ancestor. Two ancestral species with nine pairs of chromosomes, S and L, mate to produce a hybrid S/L offspring with 18 chromosomes that do not pair. This sterile offspring undergoes whole genome duplication to produce a new fertile species with 18 pairs of chromosomes that is ancestral to all Xenopus species. For clarity, the process is rendered here with only one type of chromosome. Copyright: 2016 Masanori Taira. |
Scientists believe that two rounds of whole genome duplication 500 million years ago contributed to the emergence of the first vertebrate species and an explosion of diversity in this group. The new findings from the X. laevis genome should improve our understanding of this important event in our evolutionary history.
“I’m surprised that our results indicate that the subgenomes of X. laevis are just evolving now,” said Akimasa Fukui. “I believe that these data will not only help us understand the evolutionary aspect of vertebrates, but through frog research, also lead to applications in regenerative therapy.”
Original article:
Adam M. Session, Yoshinobu Uno, Taejoon Kwon, et al., Genome evolution in the allotetraploid frog Xenopus laevis, Nature, October 19, 2016. DOI: 10.1038/nature19840
Funding information:
U.S. National Institute of Child Health and Human Development through grants HD065705, HD080708, P41 HD064556, GM086321 DSR, RMH AMS, JBL.TM, SM and P41 HD064556 to Xenbase; Japan Society for the Promotion of Science KAKENHI Grant Numbers, 221S0002 (A.T., Y.K., A.F.), 24590232 (S.T.), 25440180 (A.H.), 15K14521 (M.K.), 15K07082 (H.Ogino), 22570137 (T.T), 25460245 (A.S.), 23370059 (Y.I.), 23113004 (Y.M.), 25251026 (M.T.), and 22127007 (N.U.). Additional support was provided by the UNIST Research Fund Grant Number 1.150094.01, 1.150043.01 and 1.160060.01 (T.K), U.S. National Human Genome Research Institute (J.S.), the Hiroshima University Phoenix Leader Education Program (S.T.), National BioResource Project of MEXT Japan (A.S.), the US National Institute of Health Office of the Director and US National Institute of Allergy and Infectious Diseases (M.F.F., Y.O.), the US National Institute of General Medical Science (I.Q., S.H., J.B.W.), the US National Science Foundation (NSF), Cancer Prevention Research Institute of Texas(CPRIT) and Welch(F1515) (E.M.M.), the US NIHGM and NHLBI (J.B.W.), and the Okinawa Institute for Science and Technology Graduate University (O.S). Work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 DSR AMS, JAC, UH, SS, JC, JJ, JG, JS; J.K. was supported by an NSF graduate research fellowship, and A.M.S. was supported by a NHGRI Training Grant. Grant R01HD069344 to GJCV.
Contacts:
Associate Professor Akimasa Fukui
Faculty of Advanced Life Science
Laboratory of Tissue and Polymer Sciences
Hokkaido University
Email: afukui[at]sci.hokudai.ac.jp
Naoki Namba (Media Officer)
Global Relations Office
Institute for International Collaboration
Hokkaido University
Email: pr[at]oia.hokudai.ac.jp
Tel: +81-11-706-8034